なんとか2月の3講演のスライドを完成させ、和文総説も前倒しで終えました。ガイドラインの資料もほぼ完成して、委員のチェックが終わればパブコメです。
今回の論文チェックは 1ヶ月分だから楽かなとおもったら、面白い論文が多すぎて、2回に分けました。
①Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis. (2016.12.21 published online)
多発性硬化症 (一次性進行型を除く) に対して、オクレリズマブとインターフェロンβ1aを比較した 2つの第三相試験 (OPERA I/II) をまとめた報告。Double-dummyで double-blindとのこと。オクレリズマブは、CD20陽性 B細胞に対するモノクローナル抗体である。オクレリズマブ群は、600 mgを 24週間毎に経静脈投与した。一方、インターフェロンβ1a群は 44 ug週 3回の皮下注射を 96週間続けた。その結果、主要エンドポイントである年間再発率は OPERA I試験でオクレリズマブ群 0.16, インターフェロンβ1a群 0.29 (オクレリズマブ群で 46%低い, p<0.001) で、OPERA IIでオクレリズマブ群 0.16, インターフェロンβ1a群 0.29 (オクレリズマブ群で 47%低い, p<0.001) であった。両試験を併せると、12週間後に障害が進行した患者の割合は、オクレリズマブ群でインターフェロンβ1a群に比べて有意に低かった (9.1 v.s. 13.6%, hazard ratio 0.60, 95%信頼区間 0.45~0.81, p<0.001)。また24週間後についても同様であった (6.9 v.s. 10.5%, hazard ratio 0.60, 95%信頼区間 0.43~0.84, p=0.003)。ガドリニウム造影病変の平均個数は、オクレリズマブ群で 0.02個、インターフェロンβ1a群で 0.42個であった。注射関連反応がオクレリズマブ群の 34%でみられた。重症感染は、オクレリズマブ群 1.3%で、インターフェロンβ1a群 2.9%であった。悪性腫瘍はオクレリズマブ群 0.5%で、インターフェロンβ1a群 0.2%だった。
ベースラインで半数以上の患者の EDSSが 0という、比較的軽症者を対象とした試験。経静脈投与の薬剤の方が、皮下注射で用いるインターフェロンなどより効果が高いというのは、他の薬剤でもそうなので驚くような結果ではない。むしろ驚いたのは次の文章。スポンサーとズブズブの臨床試験じゃないか・・・。”The sponsor, F. Hoffmann–La Roche, designed the trial in consultation with members of the ORATORIO trial steering committee. Data were collected by the site investigators, queries were responded to by site personnel, and the data were analyzed by the sponsor; the aggregated and individual results of the participants were reviewed by the sponsor and steering committee.”, “A subgroup of authors, which included academic authors and authors who are employees of the sponsor, drafted the manuscript, and all the authors approved the final version and made the decision to submit the manuscript for publication. Medical-writing assistance was funded by the sponsor.” という記載。あと、注射関連反応が 34%にみられた (一方でインターフェロンβ1aは10%) ということは、そこで盲検化が崩れたりはないのだろうか・・・。
②Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. (2016.12.21 published online)
多発性硬化症の一次進行型を対象に、オクレリズマブとプラセボを比較した第三相試験。732名の一次進行型多発性硬化症患者を、2:1でオクレリズマブ 600 mgかプラセボに割り付け。薬剤の経静脈投与は 24週間ごとに、少なくとも 120週間もしくは事前に定められた数の障害進行イベントが起こったときまでとした。主要エンドポイントは 12週間の時点で障害の進行が確認された患者の割合とした。その結果、12週での障害が進行した患者の割合は、オクレリズマブ群 32.9%で、プラセボ群 39.3%だった (Hazard ratio 0.76, 95%信頼区間 0.59~0.98, P=0.03)。24週ではオクレリズマブ群 29.6%で、プラセボ群 35.7%だった (Hazard ratio 0.58, 95%信頼区間 0.58~0.98, P=0.04)。120週での timed 25-foot walk (25フィート歩行の時間) の悪化は、オクレリズマブ群 38.9%で、プラセボ群 55.1%だった (P=0.04)。T強調像での病変は、オクレリズマブ群で 3.4%減少に対し、プラセボ群では 7.4%増加した。
一次進行型の多発性硬化症に効果が証明された薬剤は存在しないので、もし有効であれば凄いことだと思う。同じく CD20阻害薬であるリツキシマブは、有効性を証明できなかった (OLYMPUS trial) とのこと。この臨床試験も、同じ号に掲載されたインターフェロンβ1aとの比較同様、次の文句が並ぶ。”The sponsor, F. Hoffmann–La Roche, designed the trial in consultation with members of the ORATORIO trial steering committee. Data were collected by the investigators and analyzed by the sponsor; the results were reviewed by the sponsor and steering committee.” “The first draft of the manuscript was written by the first and last authors, with medical writing assistance funded by the sponsor.”
③An observational study of alemtuzumab following fingolimod for multiple sclerosis. (2017.1.10 published online)
フィンゴリモドを中止して 12ヶ月以内にアレムツズマブを開始すると、多発性硬化症の疾患活動性が高くなることを示唆する 9症例の報告。アレムツズマブに先行してフィンゴリモドが投与されていた 36例中 9例なので、実は多いのかもしれない。
多発性硬化症の新薬は次々と生まれていて、組み合わせ次第では様々なことが起きるのではないかという気がする。これは氷山の一角ではないだろうか。
④Severe B-cell-mediated CNS disease secondary to alemtuzumab therapy. (2017年2月号)
41歳男性にアレムツズマブの初回投与を行った後、症状の急激な増悪があり、頭部MRIで造影される新規病変が 20ヶ所出現していた。メチルプレドニゾロン 7000 mgの静脈内投与を行ったが改善なく、免疫吸着療法が奏功した。病勢を安定させるため、リツキシマブを追加した。その後、症状は安定し、新規病巣の出現もない。
アレムツズマブを使う機会はそうそうないけれど、こういうこともあるのかと。しかし、そこでリツキシマブを使うとは、かなり勇気が必要だっただろうなぁ・・・。
⑤Soluble CD27 Levels in Cerebrospinal Fluid as a Prognostic Biomarker in Clinically Isolated Syndrome. (2017.1.3 published online)
臨床的イベントが 1回のみで、まだ多発性硬化症と呼べないものを clinically isolated syndromeと呼ぶが、将来多発性硬化症になる患者ではそうでない患者と比べて髄液中の可溶性 CD27が高いという報告。
確かにそうかもしれないけれど、個々の症例で将来多発性硬化症を発症するかどうかの予測に使えるほどはっきり分離できる結果ではないと思った。
⑥MOG antibody-positive, benign, unilateral, cerebral cortical encephalitis with epilepsy. (2017.1.16 published online)
東北大学からの報告。てんかんを伴う片側性皮質性脳炎で抗MOG抗体を測定したところ陽性であった。そこで、24例の原因不明の成人ステロイド反応性脳炎で抗MOG抗体を測定した。その結果、さらに 3名が抗MOG抗体陽性であった。全例男性で、年齢中央値 37歳 (23~39歳)、主要な症状は全般性てんかん発作であった。2例では片側性の良性視神経炎があった。全例、頭部MRIで片側の大脳皮質が FLAIRで高信号となっており、それらは腫脹し、SPECTでの血流増加に合致した所見であった。髄液では、単核球優位の中等度の細胞数増多と軽度の蛋白上昇があったが、ミエリン塩基性蛋白の上昇はなかった。他の抗体は陰性で高用量のメチルプレドニゾロンが有効で、MRIでの病巣は消失した。再発はなかった。
頭部MRIで片側皮質の FLAIR高信号を伴う自己免疫性脳炎では、抗MOG抗体の測定が必要・・・というのは勉強になった。
⑦Anti-LGI1 encephalitis is strongly associated with HLA-DR7 and HLA-DRB4. (2016.12.27 published online)
悪性腫瘍非合併の抗LGI1抗体による脳炎患者 25名の HLAを調べたところ、HLA-DR7が 88% (コントロール群 19.6%)、HLA-DR4が 100% (コントロール群 46.5%) であった。著者らは、HLA-DR7や HLA-DR4を欠く抗LGI-1抗体脳炎では、悪性腫瘍の疑いがあるかもしれないとしている。
⑧Characteristics in limbic encephalitis with anti-adenylate kinase 5 autoantibodies. (2017.1.6 published online)
抗AK5抗体陽性脳炎 10例の纏め。平均年齢中央値 64歳 (57~80歳)、けいれん発作を伴わない前向性健忘、ときどき無力症 (asthenia) や気分障害の先行がある。癌との関連はない。全例、MRIで両側の海馬以上信号があった。髄液検査では、軽度の細胞数増多、IgG index上昇、オリゴクローナルバンド、タウ上昇 (Aβやリン酸化タウは正常) がみられた。1例を除き、免疫療法は効果がなく、高度な前向性健忘が残存した。2例では重度の認知機能低下が進行した。MRIでその後海馬萎縮がみられた。In vitroでは 抗AK5抗体の役割を同定することはできなかった。
高齢者の前向性健忘を伴う脳炎で、当初 FLAIRで海馬の異常信号があり、やがて萎縮するという、似たような症例の経験がある。この報告と異なっていたのは、てんかんを合併していたこと。昔働いていた病院なのだけど、この疾患だったのかどうか気にかかる。
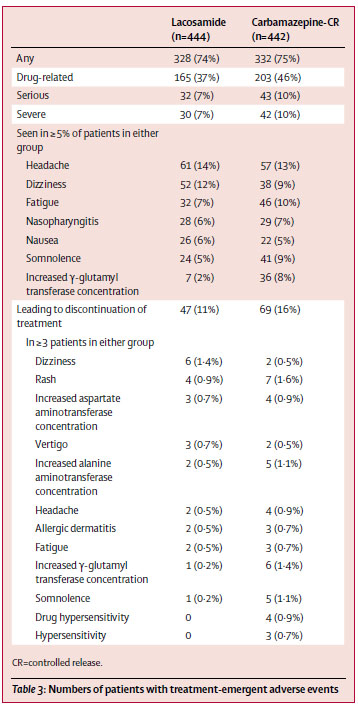
⑨Efficacy, safety, and tolerability of lacosamide monotherapy versus controlled-release carbamazepine in patients with newly diagnosed epilepsy: a phase 3, randomised, double-blind, non-inferiority trial. (2016.11.24 published online)
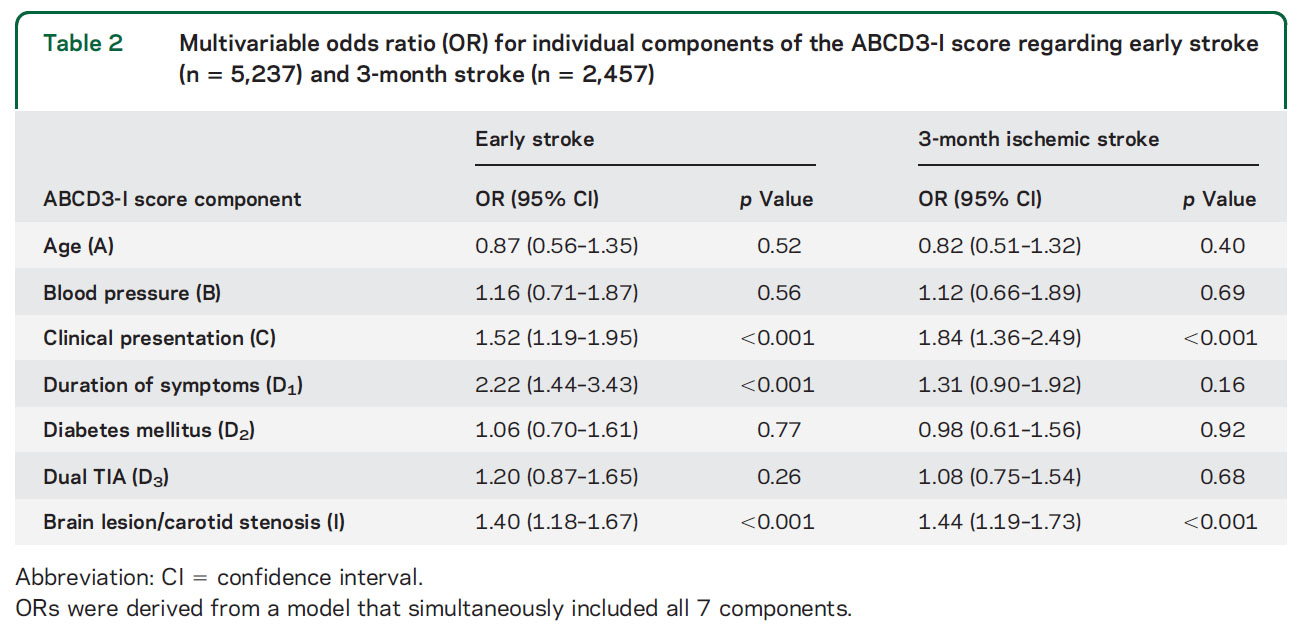
新規抗てんかん薬ラコサミドの第三相試験。16歳以上の新規てんかん患者が対象。介入群はラコサミド (100→200 mg/day, 発作があれば max 600 mg/dayまで増量)、対照群はカルバマゼピン徐放錠 (200→400 mg/day, 発作があれば max 1200 mg/dayで増量)。主要エンドポイントは 6ヶ月連続で発作消失。6ヶ月の治療を継続できたのは、ラコサミド群 74% (327名)、カルバマゼピン徐放錠群 70% (308名) だった。6ヶ月間の発作抑制は、ラコサミド群 90%、カルバマゼピン徐放錠群 91%で、絶対リスク差 -1.3% (95%信頼区間 -5.5~2.8%)、だった。ラコサミドはカルバマゼピン徐放錠に非劣性だった。
有害事象の総数は両群でそれほど大きな違いはないが、ラコサミド群では浮動性めまいが多く、カルバマゼピン徐放錠では倦怠感、皮疹、傾眠、肝機能障害が多い傾向にあった。
副作用
⑩Monotherapy treatment of epilepsy in pregnancy: congenital malformation outcomes in the child. (2016.11.7 published online)
妊婦への抗てんかん薬単剤療法の Cochrane reviewが公開されている。包含基準を満たした 50の臨床試験のうち、31試験で meta-analysisをおこなった。その結果、大奇形のリスクが最も低かったのはレベチラセタムとラモトリギンであった (キモの部分は “There was no increased risk for major malformation for lamotrigine (LTG). Gabapentin (GBP), levetiracetam (LEV), oxcarbazepine (OXC), primidone (PRM) or zonisamide (ZNS) were not associated with an increased risk, however, there were substantially fewer data for these medications.For AED comparisons, children exposed to VPA had the greatest risk of malformation (10.93%, 95% CI 8.91 to 13.13).”)。しかし、特異的な奇形についてのデータには乏しく、内科医はリスクと治療の有効性について、患者とよく話し合うべきである。(日本語要約)
薬剤選択について従来の結論と大きな違いはないが、こういう質の高い研究で示された意義は大きい。さまざまな抗てんかん薬の直接比較をおこなっているのだが、抗てんかん薬は種類が多く、比較には様々な組み合わせがあるため、解析を見ると Comparison 66までおこなっていて、大変な努力が必要な研究だと思った。
⑪Rhabdomyolysis Associated with Levetiracetam Administration. (2016.12.31 published online)
レベチラセタム静脈内投与後に横紋筋融解症をきたした 27歳女性の報告。レベチラセタム単剤で治療を始めた後に出現し、無症候性で血清 CKの最大値は 49539 U/lだった。これまでレベチラセタムに関連した横紋筋融解症は 4例報告されている。全例 13~29歳で、CKは 986~29136 U/lだった。4名中 3名では併用薬があり、単剤だった 1名の CKは 986 U/lだった。
よく使う薬剤なので、極めて稀ではあっても、知っておきたい副作用。
⑫Glucocorticoid-associated worsening in reversible cerebral vasoconstriction syndrome. (2017.1.17 published online)
可逆性脳血管攣縮症候群は、繰り返す雷鳴頭痛を呈し、約 1/3~1/2に虚血や出血を合併する。中枢神経原発血管炎 (primary angiitis of the CNS; PANCS) との鑑別を要する。mRS 3点を超えるアウトカム不良は 5~14%とされている。著者らは、後方視的に臨床的あるいは画像上の増悪をきたす要因を調べた。その結果、ステロイドを投与された患者の 37%で臨床的な悪化があった (ステロイド非投与では 5%)。また、臨床的悪化のあった患者で退院時の mRS 4-6の予後不良の割合は、ステロイド投与群で ステロイド投与群 47%、非投与群 17%だった。ベースラインの重症度で、ステロイド投与群が非投与群より軽症であったわけではなく (むしろベースラインの画像ではステロイド投与群の 35%が正常で、非投与群では全例異常があった)、ステロイド投与から症状増悪まで、ほとんどが 2日以内だった。SSRIで症状や画像上の悪化がみられたが、退院時の障害スコアへの影響はなかった。カルシウム拮抗薬やトリプタン製剤は、頭痛や増悪の予防に効果はなかった。
後方視的な研究だが、かなり説得力のある報告である。PANCSだった困るという理由で安易にステロイドを使ってしまうと、予後を悪化させる恐れがあるというのは、臨床医にとってジレンマとなるかもしれない。
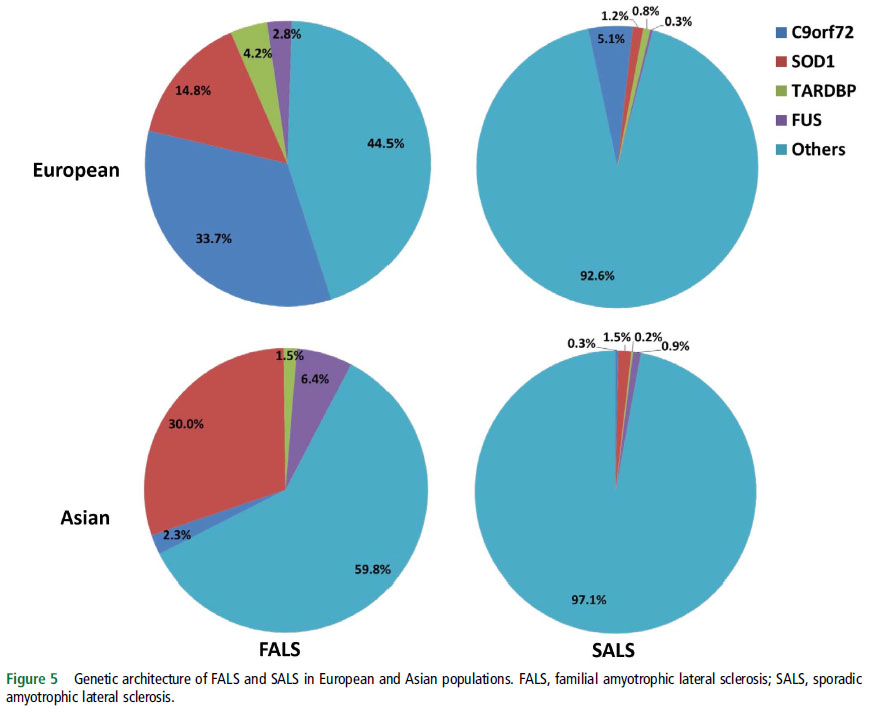
⑬Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. (2017.1.5 published online)
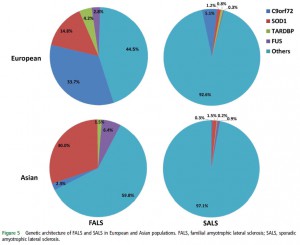
筋萎縮性側索硬化症 (ALS) の遺伝子についての systematic reviewおよび meta-analysisをおこない、ヨーロッパ人とアジア人を比較している。ヨーロッパ人では C9orf72変異が多く、日本人では SOD1変異が多い。
それぞれの頻度については、図を見ると一目瞭然。
Genetic epidemiology of ALS
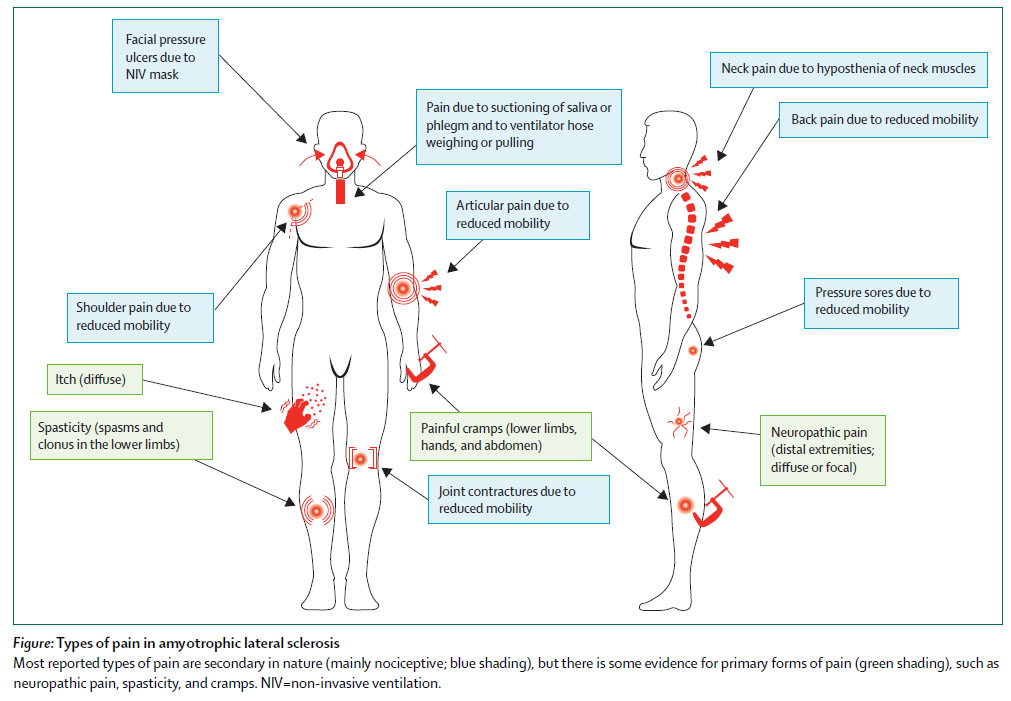
⑭Pain in amyotrophic lateral sclerosis. (2016.12.8 published online)
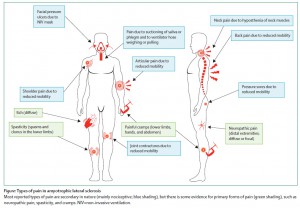
筋萎縮性側索硬化症 (ALS) と疼痛についての総説。ALSでは様々なタイプの疼痛がある。頻度については研究によりばらつきが大きい。軽度のことが多いが、中等度以上の疼痛もみられる。メカニズムとしては、神経障害性疼痛、侵害受容性疼痛、中枢性感作がある。治療法について、2013年のコクランレビューでは RCTなしとしている。著者らは、疼痛で用いる薬剤を次のように推奨している。
・神経障害性疼痛 (四肢):Gabapentin (First line), Pregabalin (First line), 三環系抗うつ薬 (First line)
・筋痙攣 (下肢、手、腹部):Quinine sulphate (First line), Gabapentin (Second line), Mexiletine (First line), Dronabinol (Second line), Levetiracetam (First line)
・痙性 (下肢):Levetiracetam (First line), Baclofen髄注 (Second line),Baclofen内服/tizanidine/dantrolene/benzodiazepines (NA)
・体動困難に起因する疼痛:NSAIDs and paracetamol (First line), Opioids (Second line), Cannabis (Second line)
・四肢の麻痺 (肩痛, 関節痛):リドカイン・ステロイドの関節内注射 (First line)
ALSと疼痛のタイプ
ALSで疼痛を訴える患者さんはたまにいるので読んでみたけれど、やはりわかっていないことが多いみたい。治療薬としては NSAIDs/アセトアミノフェンもしくは神経障害性疼痛についての薬剤がメインになるというのは予想通り。
【後編】へ続く